
Lyzozómové ochorenia ako príčina dyslipoproteinémie
Lysosomal storage disorders as the cause of dyslipoproteinemia
Lysosomal storage disorders (LSDs) belong to group of rare, inborn, inherited diseases. They are caused by insufficient activity of certain lysosomal enzymes, by error of a transport protein or by malfunction of an enzymatic activator. Onset of symptoms occur anytime from infancy until adulthood. Early onset forms of disorders are commonly linked with severe natural course, quick progression of organ failure and with generally poor prognosis. Late onset of disorder, during adulthood, is commonly linked with mild form of disease. Without appropriate therapy they also lead to sever organ failure. Natural course of these disorders is multisystemic with progression of clinical impairments. The most affected are metabolic active tissues and organs: bone marrow, liver, bones, muscles, myocardium or central nervous system. Although dyslipoproteinemia is a common finding during routine biochemical screening it is often overseen and can prolong the time needed for establishing the right diagnosis. Authors of this paper are discussing some of the LSD connected with dyslipoproteinemia, which are currently treatable. They are describing clinical presentation, diagnostic procedures and actual therapeutic approaches of inborn errors of metabolism.
Key words:
diagnostic approach, dyslipoproteinemia, enzyme replacement therapy, lysosomal storage disorders, substrate reducing therapy
Received: 24. 9. 2018
Accepted: 1. 10. 2018
Autoři:
Anna Hlavatá; Katarína Juríčková
Působiště autorů:
Detská klinika LF UK a NÚDCH, Centrum dedičných metabolických porúch, Bratislava
Vyšlo v časopise:
AtheroRev 2018; 3(3): 184-191
Kategorie:
Přehledové práce
Souhrn
choroby spôsobené nedostatočnou aktivitou niektorého z lyzozómových enzýmov, poruchou transportného proteínu či enzýmového aktivátora. Prvé príznaky ochorenia sa môžu objaviť kedykoľvek od novorodeneckého obdobia až po dospelosť. Včasné formy mávajú ťažký priebeh s rýchlou progresiou a infaustnou prognózou. V dospelosti môže mať začiatok choroby miernejší priebeh, ktorý však bez liečby ústi do závažného ochorenia. Postihnutie je multisystémové s trvalou progresiou klinických ťažkostí, s postihnutím najmä metabolicky aktívnych tkanív a orgánov: kostná dreň, pečeň, kosti, kostrové svalstvo, myokard či centrálny nervový systém. Pri úvodnom biochemickom vyšetrení krvi pacienta môže byť zistená dyslipoproteinémia, ktorá ak nie je posudzovaná spolu s ďalšími klinickými príznakmi u pacienta, môže oddialiť stanovenie správnej diagnózy. Autori vo svojej práci poukazujú na niektoré lyzozómové ochorenia spojené s dyslipoproteinémiou, ktoré už dnes patria medzi liečiteľné choroby. Popisujú klinický obraz, dostupnú diagnostiku a aktuálnu liečbu týchto vrodených metabolických porúch.
Kľúčové slová:
diagnostika, dyslipoproteinémia, enzýmová substitučná terapia, lyzozómové choroby, substrát redukujúca terapia
Úvod
Lyzozómové ochorenia (Lysosomal Storage Disorders – LSDs) sú geneticky podmienené poruchy degradácie makromolekúl väčšinou na podklade nedostatočnej aktivity lyzozómových enzýmov. Príčinou však môže byť aj nesprávna funkcia lyzozómového transportného proteínu či enzýmového aktivátora. V dôsledku deficitu či nedostatočnej funkcie týchto kľúčových proteínov dochádza k hromadeniu (tezaurácii – odtiaľ starší názov chorôb tezaurizmózy) produktov metabolizmu v organizme. Tvoria heterogénnu skupinu v rámci dedičných metabolických ochorení. V dnešnej dobe je popísaných viac ako 50 rozličných nozologických jednotiek. Dedičnosť je majoritne autozómovo recesívna, výnimku tvoria X-viazané ochorenia – Fabryho choroba, mukopolysacharidóza typ II, Danonova choroba, či autozómovo dominantne dedičná adultná forma neuronálnej ceroidnej lipofuscinózy. Diagnózu lyzozómového ochorenia definitívne potvrdíme stanovením zníženej enzýmovej aktivity daného enzýmu, v prípadoch poruchy nekatalytických proteínov je potrebné molekulovo-genetické vyšetrenie. Nadobúdaním nových poznatkov o biochemicko-patomechanickej podstate jednotlivých porúch a rozvojom výskumu zameraného na pomoc pacientom s týmito ochoreniami, sa podarilo niektoré z nich označiť za terapeuticky ovplyvniteľné.
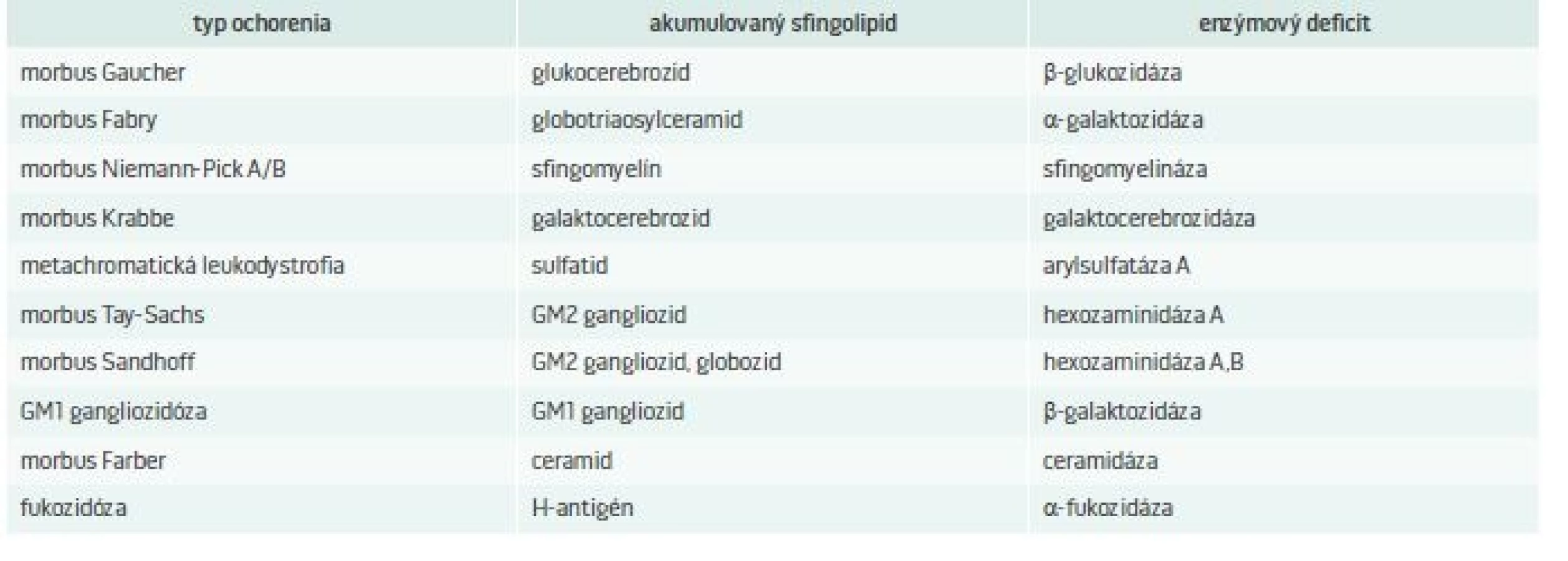
Lyzozóm je sférická membránová organela o veľkosti 0,1–1,2 μm. Bola objavená v roku 1955 belgickým cytológom Christianom de Duve [1]. Názov pochádza z gréckeho slova lysis, čiže rozpustenie, zničenie a slova soma, teda telo. Lyzozómy sú prítomné vo všetkých bunkách organizmu okrem erytrocytov. Lyzozóm je zapojený do katabolizmu bunkového materiálu, ktorý sa do neho dostane pomocou autofágie [2]. Objavy za posledné desaťročia pomohli objasniť úlohu lyzozómu nielen ako organely s významnou úlohou pri degradácii materiálu, ale aj ako dôležitého hráča v regulácii viacerých bunkových funkcií. Narušená funkcia lyzozómu spôsobuje nerovnováhu bunkovej homeostázy a vedie k lyzozómovým ochoreniam [3]. Základné rozdelenie LSDs je zhrnuté v tab.1.

Dyslipoproteinémia pri LSDs
LSDs, pri ktorých dochádza typicky k akumulácii komplexných tukov v jednotlivých bunkách a tkanivách, čo vedie k poruche lipidovej homeostázy, nazývame tiež lipidózy. Medzi tieto ochorenia patria najmä sfingolipidózy, Niemannova-Pickova choroba typ C a deficit kyslej lyzozómovej lipázy [4].
Poruchy metabolizmu sfingolipidov nie sú vyvolané ich abnormálnou syntézou, ale chybnou degradáciou.
Sfingolipidózy tvoria významnú skupinu v rámci LSDs. Sfingolipid (schéma 1) vo svojej molekule obsahuje aminoalkohol s dlhým reťazcom (sfingozín), ktorý sa spája s mastnou kyselinou a vytvára ceramid čiže zložený lipid. Glykosfingolipidy sú deriváty ceramidov: naviazaním sacharidového zvyšku na ceramid vzniká cerebrozid. Tie sú významné pri stavbe myelínových pošiev a slúžia ako prekurzory pre zložitejšie glykosfingolipidy, napríklad sulfatidy po naviazaní sulfátovej skupiny. Pripojením polysacharidu na ceramid vzniká globozid. V prípade pripojenia jedného alebo viacerých zvyškov kyseliny N-acetylneuramínovej (sialovej kyseliny) na globozid vnikne gangliozid. Fosfosfingomyelíny (sfingomyelíny) sú taktiež deriváty ceramidov: na sfingozín je naviazaná kyselina fosforečná s cholínom. Fosfosfingolipidy patria medzi najrozšírenejšie sfingolipidy v živočíšnych tkanivách. Sú súčasťou mozgovej bielej hmoty a tvoria myelínové pošvy nervov.
![Schéma 1 | Štruktúra sfingolipidu: sfingozín spolu s mastnou kyselinou vytvára ceramid. Po naviazaní monosacharidu
na ceramid vzniká glukocerebrozid. V prípade naviazania ceramidu s polysacharidom a s jedným alebo
viacerými zvyškami kyseliny N-acetylneuroaminovej je výsledkom gangliozid. Upravené podľa [5]](https://pl-master.mdcdn.cz/media/image_pdf/2ee84d1a6c46ef8839dc1cef02307ecd.jpeg?version=1540462580)
LSDs, pre ktoré je charakteristické ukladanie sfingolipidov a enzýmový deficit, ktorý ich spôsobuje, uvádza tab. 2. Pri týchto ochoreniach dochádza k akumulácii rôznych lipidov, ako sú ceramidy, cerebrozidy, sulfatidy, sfingomyelíny, gangliozidy a lipofuscíny [5].
Je známe, že lipoproteín s vysokou hustotou (HDL) má protektívny efekt v rámci aterogenézy, čím znižuje kardiovaskulárne riziko. Dôležitou funkciou HDL je úloha pri tzv. reverznom transporte cholesterolu, pri ktorom HDL prenáša cholesterol z periférnych tkanív do pečene na jeho elimináciu.
Apolipoproteín A1 (apoA1) je hlavnou štrukturálnou súčasťou HDL-cholesterolu. V spolupráci s ABCA1 transportérom (ATP-binding cassette transporter A1) sprostredkováva odchod prebytočného cholesterolu a sfingolipidov z bunky, čo je prvý krok v procese reverzného transportu cholesterolu [6]. Mechanizmus, akým prebieha regulácia interakcie medzi apoA1 a ABCA1 nie je jednoznačný. ABCA1 sa nachádza nielen na plazmatickej membráne, ale aj v endocytických vezikulách, ktoré premávajú medzi neskorými endocytickými kompartmentami a bunkovým povrchom. Predpokladá sa, že ABCA1 v neskorých endocytických vezikulách, t.j. v neskorom endozóme a v lyzozóme, mobilizuje lipidy pre apolipoproteín A1 (apoA1), čím sprostredkováva ich eflux z bunky. ApoA1 sa naviaže na bunkový povrch a vchádza do bunky spolu s ABCA1 v podobe skorého endozómu. Časť ABCA1 a apoA1 prichádza až do neskorého endozómu a lyzozómu. ABCA1 nachádzajúci sa na bunkovom povrchu ako aj v skorých endocytických vezikulách zabezpečujú lipidáciu (proces, pri kotorom je na molekulu proteínu naviazaná lipidová skupina) apoA1. ApoA1 prichádza k bunkovému povrchu naspäť, kde je uvoľnený ako nascentná molekula HDL [7].
Viaceré štúdie potvrdili, že interakcia medzi glykosfingolipidmi a cholesterolom spôsobuje jeho akumuláciu vo vnútri bunky. Cholesterol ako aj iné lipidy sa akumuluje primárne či sekundárne pri viacerých LSDs. Predpokladá sa, že akumulácia nedegradovaného materiálu zabraňuje prepravovaniu v rámci endozómovo-lyzozómového systému. ApoA1 mediovaný eflux cholesterolu je sprostredkovaný cez ABCA1 (ATP-Binding cassette transporter), ktorý je kľúčovým regulátorom bunkovej homeostázy cholesterolu. Akumulácia glykosfingolipidov narúša a inhibuje odchod cholesterolu cez apoA1 [8]. Narušená homeostáza lipidov v endotelových bunkách spôsobuje stimulácia proinflamačných molekúl, čím urýchľuje aterosklerózu [9].
Nižšia koncentrácia HDL-cholesterolu, ktorá sa spája s vyšším kardiovaskulárnym rizikom, bola pozorovaná u pacientov s Gaucherovou chorobou [10] a Niemannovou-Pickovou chorobou typu A/B či C (NP A/B, NPC). U pacientov s Fabryho chorobou bola koncentrácia HDL v norme, resp. zvýšená [11].
Niemannova-Pickova choroba typ C nepatrí medzi enzymopatie. Je charakteristická akumuláciou neesterifikovaného cholesterolu v bunke, ktorá je spôsobená mutáciou v géne pre transportné proteíny NPC1 alebo NPC2. Oba tieto proteíny sa nachádzajú v endozómovo-lyzozómovom systéme a spolupracujú na odbúravaní cholesterolu. Narušená funkcia týchto proteínov vedie k poruche transportu cholesterolu z endozómovo-lyzozómového systému. Akumulácia cholesterolu vedie k lipidovej nerovnováhe bunky. Dochádza k sekundárnej akumulácii sfingomyelínov, neutrálnych glykolipidov či gangliozidov [4, 12].
Pri deficite enzýmu kyslej lyzozómovej lipázy (Lysosomal Acid Lipase Deficiency – LAL-D) dochádza k poruche intralyzozómovej degradácie triacylglycerolov a esterov cholesterolu. LAL štiepi LDL derivovaný ester cholesterolu na neesterifikovaný cholesterol [13]. Neesterifikovaný cholesterol má významnú úlohu pri endogénnej syntéze cholesterolu. Pri deficite LAL sa estery cholesterolu akumulujú v lyzozómovom systéme najmä makrofágov pečene a sleziny [14, 15], ale tiež v hepatocytoch a bunkách kôry nadobličiek. Pri laboratórnom vyšetrení je u pacientov trvalá hypercholesterolémia s vysokou hodnotou LDL-C spôsobená zvýšenou syntézou VLDL hepatocytmi, nízka hodnota HDL-C a variabilne zvýšené triacylglyceroly [16].
Diagnostika lyzozómových chorôb
Diagnostika LSDs sa odvíja od typickej klinickej symptomatológie, ktorá spolu s laboratórnymi a zobrazovacími vyšetreniami vyústi do biochemicko-metabolických testov, enzymologického a molekulovo-genetického vyšetrenia.
Po zhodnotení klinického obrazu, anamnestických údajov, výsledkov tzv. bežných laboratórnych (krvný obraz, náter periférnej krvi, biochemické parametre) a zobrazovacích vyšetrení (RTG, ultrasonografia parenchymatóznych orgánov, magnetická rezonancia mozgu), sú pokračujúcimi vyšetreniami špeciálne metabolické screeningové vyšetrenia. Diagnostika lyzozómových enzymopatií je finalizovaná enzymologickým a molekulovo-genetickým vyšetrením [17,18], pri poruche nekatalytických proteínov je definitívne potvrdená diagnóza molekulovo-genetickým vyšetrením, ako je to napríklad pri Niemannovej-Pickovej chorobe typu C. Výrazný prelom v diagnostike lyzozómových enzymopatií nastal možnosťou stanovenia enzýmovej aktivity zo suchej kvapky kapilarizovanej krvi (SKK) nabratej na filtračný papierik. Táto metodika výrazne posunula vpred diagnostiku LSDs. Postupne sa pomocou suchej kvapky krvi dajú diagnostikovať pacienti s liečiteľnými formami mukopolysacharidóz (MPS I,II, IVA,VI,VII), Pompeho, Fabryho a Gaucherovou chorobou aj LAL-D. Novodobé diagnostické metódy umožnili pri LSDs ústup od invazívnych vyšetrení s odberom tkanív k histochemickému vyšetreniu. Biopsia orgánov si však ponecháva svoje miesto v nejednoznačných prípadoch. Jedná sa často o prípady s nejasnou patogenitou nájdených mutácií pri molekulovo-genetickom vyšetrení.
Terapia lyzozómových chorôb
Lyzozómové choroby patria medzi dedičné metabolické poruchy, ktoré je možné terapeuticky ovplyvniť. Pri niektorých z nich, ktoré sú spôsobené deficitom enzýmu, je možné chýbajúci enzým nahradiť transplantáciou kostnej drene či farmakologickou substitúciou enzýmu (z angl. enzyme replacement therapy – ERT). Transplantácia kostnej drene má byť vykonaná do 24. mesiaca života dieťaťa, aby bol efektívne chránený centrálny nervový systém (CNS) [19]. Cestu k úspešnej farmakologickej liečbe náhradou chýbajúcich enzýmov otvorilo objavenie manózo-6-fosfátových receptorov makrofágov, ktoré sprostredkúvajú rozpoznanie a transport špecifických enzýmov do lyzozómov buniek. Pri ERT má pacient intravenóznou infúziou podávaný chýbajúci enzým, ktorý je vyrobený rekombinantnou technológiou. Jedná sa o veľkú molekulu–proteín, ktorý neprechádza cez hematoencefalickú bariéru, a tak nemôže ovplyvniť postihnutie CNS [20, 21]. Ďalšou možnosťou liečby pacientov s LSDs je substrát redukujúca terapia (SRT). Jedná sa o malé molekuly, ktoré blokujú syntézu nedegradovateľného materiálu, čím znižujú jeho akumuláciu. Majú schopnosť prenikať do CNS a teoreticky ovplyvniť aj neurologické ťažkosti [22, 23]. Pri niektorých LSDs je možné využiť liečbu chaperónmi, čo sú malé molekuly, ktoré väzbou na endogénny (ale i exogénne podávaný) enzým stabilizujú jeho štruktúru a zvyšujú tak jeho účinnosť [24].
Morbus Gaucher
Morbus Gaucher (Gaucher Disease – GD) je choroba, pri ktorej dochádza k akumulácii glukozylceramidu v bunkách makrofágového pôvodu v dôsledku deficitu β-glukocerebrozidázy. Nedegradovaný substrát je hromadený v bunkách monocyto- makrofágového systému, čím vznikajú tzv. Gaucherove bunky, ktoré sa nachádzajú v jednotlivých tkanivách a orgánoch. Väčšinou ide o metabolicky aktívne orgány. Prvotne býva postihnutá kostná dreň, potom retikuloendotel sleziny, Kuppferove bunky v pečeni, osteoklasty, ale aj ktorýkoľvek orgán či tkanivo ako aj CNS, pľúca či oko [25]. GD je najrozšírenejšia sfingolipidóza s autozómovo-recesívnou formou dedičnosti. V židovskej populácii je choroba veľmi častá – 1 : 450–1500, prenášači 1 : 17, inak je incidencia uvádzaná podľa jednotlivých typov 1 : 40 000–100 000. Podľa orgánového postihnutia a klinických príznakov rozdeľujeme GD do 3 základných typov. Najčastejší typ 1 – non neuronopatická forma, vyskytujúci sa až v 94 %, bol nesprávne nazývaný tiež adultný typ. Prvé príznaky sa najčastejšie objavujú už počas detstva respektíve puberty. V detskom veku býva typickým príznakom neprospievanie, nižší vzrast, neskorší nástup puberty. Pacienti sa sťažujú na únavu, ľahkú tvorbu modrín, bolesti kostí, bolesti brucha. Splenomegália či kombinovaná hepatosplenomegália patria k jedným z typických príznakov. Zvýšená krvácavosť je spôsobená útlakom červenej kostnej drene ukladajúcim sa glukocerebrozidom s následným poklesom krvotvorby. Prvé bývajú postihnuté trombocyty, potom erytrocyty a leukocyty [26]. Akumulácia nedegradovaného materiálu v kostiach spôsobuje ich remodeláciu a útlak cievneho zásobenia. Dôsledkom bývajú intenzívne bolesti kostí, takzvané kostné krízy. Tento stav býva sprevádzaný celkovými prejavmi ako zvýšená teplota, leukocytóza, zvýšenie zápalovej aktivity. Kostné krízy bývajú nesprávne interpretované ako akútna osteomyelitída, avšak hemokultúra býva opakovane negatívna. Môžu vznikať patologické fraktúry. Mentálny vývoj je bez patológie. Typ 2 – akútna neuronopatická forma – je najzávažnejšou formou GD. Príznaky sa objavujú už prenatálne či perinatálne, najneskôr v prvých mesiacoch života. Patrí k nim hepatosplenomegália, kachexia, retroflexia krku, rýchlo progredujúce neurologické postihnutie v zmysle bulbárneho syndrómu, myoklonickej epilepsie či atypický očný pohyb. Kožné postihnutie má väčšinou charakter kožnej ichtyózy typu tzv. collodion-baby. Časté sú dysmorfické rysy. K úmrtiu dochádza už väčšinou do dvoch rokov [27]. Typ 3 – chronická neuronopatická forma – vyskytuje sa približne u 5 % pacientov s GD. Manifestuje sa v skorom detstve, podobnými príznakmi ako pacienti s GD typu 1. Postupne sa však rozvíja aj neurologické postihnutie. V predškolskom či školskom veku sa objavujú atypické pohyby hlavy a očí, strabizmus, nystagmus, sekundárna epilepsia, myoklonické kŕče, tremor, dystónie, spomalenie psychomotorického vývoja až demencia. U niektorých pacientov sa rozvíjajú deformity hrudníka a chrbtice, tvorí sa až gibus. U ďalších je najzávažnejšie postihnutie myokardu, chlopní aorty často s masívnymi kalcifikáciami [26,28].
Terapia
V minulosti sa využívala transplantácia kostnej drene. Niektorí pacienti boli splenektomovaní. Odstránenie sleziny sa v dnešnej dobe využíva len pri refraktérnom hypersplenizme a masívnej splenomegálii. Pre pacientov s Gaucherovou chorobou bola ako prvá dostupná enzýmová substitučná liečba (ERT). ERT je v podobe intravenózne podávaného enzýmu vyrobeného rekombinantnou technológiou. Redukuje hepatosplenomegáliu, zmierňuje postihnutie kostí, infiltráciu kostnej drene, osteopéniu a kostné bolesti, upravuje trombocytopéniu, anémiu a celkovú slabosť chorých. Najlepší efekt liečby sledujeme u pacientov bez postihnutia CNS, t. j. typ 1 [20]. U pacientov s typom 2 nie je indikovaná. Novou možnosťou liečby je aj perorálne podávaná substrát redukujúca liečba. Je indikovaná len pre dospelých pacientov [29].
Morbus Fabry
Morbus Fabry (Fabry Disease – FD) je spôsobený poruchou lyzozómovej galaktozidázy s následnou akumuláciou neutrálnych glykosfingolipidov a produktov štiepenia α-galaktosylu (GB3-globotriosylceramid) v bunkách rôznych orgánov a tkanív (obličky – postihnutie glomerulov, tubulov, myokardu, srdcovej chlopne, ganglií zadných rohov miechy, autonómny nervový systém, endotel, hladké svalstvo ciev). I keď je dedičnosť viazaná na chromozóm X, ochorenie sa môže manifestovať aj u žien „prenášačiek“, ktoré sú tak manifestnými heterozygotkami. Vyskytuje sa približne u 1 zo 40 000 mužov či 1 zo 20 000 žien s rovnakou incidenciou vo všetkých etnických skupinách. Fabryho choroba je ochorenie s pomalou progresiou, nestretávame sa s rýchle progredujúcimi letálnymi formami. Typickým klinickým príznakom v detstve je krutá bolesť akrálnych častí tela (plosky nôh, dlane), pričom akroparestézie sú provokované teplotou či fyzickou aktivitou. Chorí zle znášajú teplo, majú znížené potenie. Neskôr sa môžu objaviť angiokeratómy na koži, poškodenie rohovky (cornea verticillata), postihnutie gastrointestinálneho traktu – bolesti brucha, hnačky. Časté bývajú bolesti hlavy, vertigo, únava, tinitus či depresia. U adolescentov či mladých dospelých sa objavujú príznaky z poškodenia obličiek či myokardu [30,31]. Ide o progresívne ochorenie skracujúce dĺžku života. Vyúsťuje do renálnej insuficiencie, infarktu myokardu, mozgovej príhody či multiorgánového zlyhania.
Terapia
Terapia sa delí na nešpecifickú a špecifickú. Prvou špecifickou liečbou od roku 2001 bola enzýmová substitučná liečba (ERT), intravenóznym podávaním enzýmu získaného metódami genetického inžinierstva. Dnes je pre konkrétne mutácie ochorenia dostupná aj perorálna terapia v podobe malých molekúl-chaperónov. Génová terapia sa začala skúšať na pacientoch v januári 2017. Do nešpecifickej (symptomatickej) liečby zaraďujeme manažment jednotlivých klinických príznakov odvíjajúcich sa od postihnutých orgánov, ako sú proteinúria a postupné obličkové zlyhávanie (ACE inhibítory, sartany, dialýza, transplantácia obličiek), liečba zlyhávajúceho srdca pre hypertrofickú kardiomyopatiu a liečba arytmie, terapia bolesti, prevencia vzniku cievnej mozgovej príhody, pomôcky pri strate sluchu, v niektorých prípadoch psychiatrická terapia [32].
Morbus Niemann-Pick typ C
Morbus Niemann-Pick typ C (NPC) – nepatrí medzi lyzozómové enzymopatie, ale je spôsobený poruchou transportu cholesterolu vo vnútri bunky. Dochádza k narušeniu procesu v priebehu endocytózy LDL-cholesterolu a jeho sekvestrácie do lyzozómu. Transport z perinukleárneho lyzozómu do bunkovej membrány a endoplazmatického retikula je spomalený až zastavený. V dôsledku deficitu transportného proteínu NPC1 alebo NPC2 je cholesterol zadržaný v lyzozómoch postihnutých buniek, akumuluje sa a vedie k narušeniu ich funkcie. Cholesterol je dôležitou súčasťou bunkovej steny. Jeho akumulácia v bunke s následným nedostatkom v bunkovej stene spôsobuje dysfunkciu až apoptózu bunky. Cholesterol je potrebný napr. aj k myelinizácii nervových vlákien, v ktorých jeho deficit spôsobuje vážne poruchy. NPC1 proteín je dôležitý taktiež na transport sfingozínu, ktorý sa akumuluje spolu s cholesterolom v bunkách a prehlbuje poruchu ich funkcie [33]. Výskyt sa uvádza 1 : 120 000. Klinické príznaky ochorenia začínajú u 50 % pacientov v novorodeneckom a dojčenskom veku cholestatickou hepatopatiou a hepatosplenomegáliou, ktorá vo väčšine prípadov ustúpi behom niekoľkých mesiacov. Možný je aj nález izolovanej splenomegálie, ktorý môže aj niekoľko rokov predchádzať rozvoju ďalších klinických príznakov objavujúcich sa medzi 3. až 13. rokom života. V školskom veku môže byť prvým príznakom neobratnosť, zhoršenie prospechu, pamäte. Objavujú sa zmeny správania, porucha pohľadu nahor, progreduje ataxia, dysartria, extrapyramídová symptomatológia (dystónia) a demencia. Porucha hltania spôsobuje aspirácie spojené s častými infekciami dolných dýchacích ciest. Charakteristickým príznakom je gelastická kataplexia – náhla strata svalového tonusu vyvolaná emóciou. Môžu sa objaviť aj záchvaty kŕčov s rozvojom sekundárnej epilepsie. V dospelosti môže byť jediným symptómom psychiatrické postihnutie – schizofrénia, bipolárna porucha či demencia [34,35]. Postupný vývoj klinických príznakov a orgánového postihnutia je zobrazený na schéme 2.

Terapia
Jedinou dostupnou liečbou pre NPC je substrát redukujúca terapia, ktorá blokuje syntézu glukocerebrozidu, prekurzoru nutného na syntézu glykosfingolipidov. Tým dochádza k redukcii akumulácie nedegradovaného materiálu v bunkách. Touto špecifickou liečbou je možné zmierniť priebeh ochorenia a spomaliť jeho progresiu [22].
Ochorenie z akumulácie cholesterolových esterov
Ochorenie z akumulácie cholesterolových esterov (Morbus Wolman, cholesterol ester storage disease – CESD; deficit kyslej lyzozómovej lipázy, Lysosomal Acid Lipase Deficiency – LAL-D) je zapríčinené deficitom lyzozómovej kyslej lipázy v dôsledku čoho, ako už bolo spomenuté, dochádza k intralyzozómovej akumulácii cholesterolových esterov a triacylglycerolov. Choroba sa prejavuje vo dvoch fenotypoch: Wolmanova choroba s predpokladanou incidenciou 1 : 500 000 a ochorenie z akumulácie cholesterolových esterov s predpokladanou incidenciou 1 : 40 000. Wolmanova choroba predstavuje závažný, infantilný variant defektu s takmer nulovou enzýmovou aktivitou, s hromadením esterov cholesterolu v lyzozómoch hepatocytov, buniek kôry nadobličky, histiocytov PMS (fenazin metosulfát), enterocytov. Skoro po narodení, už v prvých týždňoch až mesiacoch života pacienta sa rozvinie vracanie, hnačky, neprospievanie. Typické klinické príznaky sú hepatosplenomegália, ikterus, črevná malabsorpcia, zväčšenie nadobličiek s kalcifikáciami a vakuolizácia lymfocytov. Často je zaostávanie v psychomotorickom vývoji. Pacienti zomierajú okolo 1 roka na hepatálne zlyhanie a kachexiu [36]. Najčastejšie sa však ochorenie manifestuje CESD fenotypom, v predškolskom veku hepatosplenomegáliou, hepatopátiou a hyperlipidémiou. Môže ústiť do fibrózy a cirhózy pečene. V dospelosti sa rozvíja predčasná ateroskleróza. Na deficit LAL treba myslieť najmä u neobéznych pacientov s nealkoholovou steatózou pečene (NAFLD) alebo s nealkoholovou steatohepatitídou (NASH).
Terapia
Pacienti majú diétu s obmedzením cholesterolu a tukov. Na úpravu dyslipidémie sa používali statíny, ktoré však nemajú dostatočný efekt na hepatálne poškodenie a progresiu fibrózy. V minulosti bola využívaná transplantácia kostnej drene či pečene. Tieto terapeutické prístupy nedosahovali očakávaný efekt a boli zaťažené vysokou mortalitou. Novou špecifickou liečbou je enzýmová substitučná liečba rekombinantným enzýmom podávaná intravenózne [37,38].
Záver
V popredí záujmu medicíny minulého storočia boli najmä časté, epidemiologicky významné choroby. Podstatne menší bol záujem o zriedkavé choroby, ktoré charakterizuje výskyt menej ako 5 chorých na 10 000 obyvateľov. Zriedkavé choroby sú ale pritom často život ohrozujúce stavy. Veľkú skupinu medzi zriedkavými ochoreniami tvoria dedičné metabolické poruchy. Od konca 20. storočia vďaka dostupnej liečbe, zlepšujúcej sa diagnostike i jej dostupnosti sa dostáva zvýšená pozornosť lyzozómovým chorobám, a to nielen medzi špecialistami na vrodené metabolické choroby, ale i z celkového pohľadu zdravotnej starostlivosti o populáciu. Dedičné metabolické ochorenia sa vo všeobecnosti spájajú najmä s pediatrickými pacientmi a s ťažkým priebehom. Postupom času, nadobúdaním nových vedomostí a vylepšovaním diagnostických metód dochádza častejšie k odhaľovaniu miernejších foriem ochorení najmä u dospelých pacientov. V dnešnej dobe existuje efektívna terapia na niektoré z týchto chorôb, preto je potrebné zachytiť čo najskôr tých pacientov, ktorí by z liečby najviac profitovali.
Doručeno do redakce: 24. 9. 2018
Přijato po recenzi: 1. 10. 2018
MUDr. Anna Hlavatá, PhD., MPH
Zdroje
- de Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol 1966; 28: 435–492. Dostupné z DOI: <http://dx.doi.org/10.1146/annurev.ph.28.030166.002251>.
- Kuehnel, W. Color Atlas of Cytology, Histology and Microscopic Anatomy. 4th ed. Thieme 2003: 34. ISBN: 3135624048.
- Settembre C, Fraldi A, Jahreiss L et al. A block of autophagy in lysosomal storage disorders. Hum Mol Genet 2008; 17(1): 119–129. Dostupné z DOI: <http://dx.doi.org/10.1093/hmg/ddm289>.
- Schulze H, Sandhoff K. Lysosomal Lipid Storage Diseases. Cold Spring Harb Perspect Biol 2011; 3(6). pii: a004804. Dostupné z DOI: <http://dx.doi.org/10.1101/cshperspect.a004804>.
- Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis 2017; 2(1–2): 1–71. Dostupné z DOI: <http://dx.doi.org/10.3233/TRD-160005>.
- Zhao G-J, Yin K, Fu Y-C et al. The interaction of ApoA-I and ABCA1 triggers signal transduction pathways to mediate efflux of cellular lipids. Mol Med Camb Mass 2012; 18: 149–158. Dostupné z DOI: <http://dx.doi.org/10.2119/molmed.2011.00183>.
- Neufeld EB, Stonik JA, Demosky SJ et al. The ABCA1 transporter modulates late endocytic trafficking: insights from the correction of the genetic defect in Tangier disease. J Biol Chem 2004; 279(15): 15571–15578. Dostupné z DOI: <http://dx.doi.org/10.1074/jbc.M314160200>.
- Glaros EN, Kim WS, Quinn CM et al. Glycosphingolipid accumulation inhibits cholesterol efflux via the ABCA1/apolipoprotein A-I pathway: 1-phenyl-2-decanoylamino-3-morpholino-1-propanol is a novel cholesterol efflux accelerator. J Biol Chem 2005; 280(26): 24515–2423. Dostupné z DOI: <http://dx.doi.org/10.1074/jbc.M413862200>.
- Schueler U, Kaneski C, Remaley A et al. A Short Synthetic Peptide Mimetic of Apolipoprotein A1 Mediates Cholesterol and Globotriaosylceramide Efflux from Fabry Fibroblasts. JIMD Rep 2016; 29: 69–75. Dostupné z DOI: <http://dx.doi.org/10.1007/8904_2015_507>.
- Zimmermann A, Grigorescu-Sido P, Rossmann H et al. Dynamic changes of lipid profile in Romanian patients with Gaucher disease type 1 under enzyme replacement therapy: a prospective study. J Inherit Metab Dis 2013; 36(3): 555–563. Dostupné z DOI: <http://dx.doi.org/10.1007/s10545–012–9529–3>.
- Stepien KM, Hendriksz CJ. Lipid profile in adult patients with Fabry disease – Ten-year follow up. Mol Genet Metab Rep 2017; 13: 3–6. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ymgmr.2017.06.010>.
- Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 2008; 9(2): 125–138. Dostupné z DOI: <http://dx.doi.org/10.1038/nrm2336>.
- Sloan HR, Fredrickson DS. Enzyme deficiency in cholesteryl ester storage disease. J Clin Invest 1972; 51(7): 1923–1926. Dostupné z DOI: <http://dx.doi.org/10.1172/JCI106997>.
- Fouchier SW, Defesche JC. Lysosomal acid lipase A and the hypercholesterolaemic phenotype. Curr Opin Lipidol 2013; 24/4): 332–328. Dostupné z DOI: <http://dx.doi.org/10.1097/MOL.0b013e328361f6c6>.
- Tadiboyina VT, Liu DM, Miskie BA et al. Treatment of dyslipidemia with lovastatin and ezetimibe in an adolescent with cholesterol ester storage disease. Lipids Health Dis 2005; 4: 26. Dostupné z DOI: <http://dx.doi.org/10.1186/1476–511X-4–26>.
- Elleder M, Poupětová H, Ledvinová J et al. Deficit kyselé (lysozomální) lipázy. Přehled českých pacientů. Čas Lék Čes 1999; 138(23): 719–724.
- Mattošová S, Chandoga J, Hlavatá A et al. Spectrum of GBA mutations in patients with Gaucher disease from Slovakia: identification of five novel mutations. Isr Med Assoc J 2015; 17(3): 166–170.
- Mattosova S, Hlavata A, Spalek P et al. Late onset form of Pompe disease. Bratisl Lek Listy 2015; 116(8): 502–505.
- Jameson E, Jones S, Wraith JE. Enzyme replacement therapy with laronidase (Aldurazyme(®)) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev 2013; (11): CD009354. Dostupné z DOI: <http://dx.doi.org/10.1002/14651858.CD009354.pub3>.
- Andersson H, Kaplan P, Kacena K et al. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics 2008; 122(6): 1182–1190. Dostupné z DOI: <http://dx.doi.org/10.1542/peds.2007–2144>.
- Starzyk K, Richards S, Yee J et al. The long-term international safety experience of imiglucerase therapy for Gaucher disease. Mol Genet Metab 2007; 90(2): 157–163. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ymgme.2006.09.003>.
- Fecarotta S, Romano A, Della Casa R et al. Long term follow-up to evaluate the efficacy of miglustat treatment in Italian patients with Niemann-Pick disease type C. Orphanet J Rare Dis 2015; 10: 22. Dostupné z DOI: <http://dx.doi.org/10.1186/s13023–015–0240-y>.
- Capablo JL, Franco R, de Cabezón AS et al. Neurologic improvement in a type 3 Gaucher disease patient treated with imiglucerase/miglustat combination. Epilepsia 2007; 48(7): 1406–1408. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1528–1167.2007.01074.x>.
- Goláň L. Migalastat v terapii Fabryho choroby. Interní Medicína Praxi 2017; 19(3): 167–170.
- Mistry PK, Cappellini MD, Lukina E et al. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am J Hematol 2011; 86(1): 110–115. Dostupné z DOI: <http://dx.doi.org/10.1002/ajh.21888>.
- Kaplan P, Baris H, De Meirleir L et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr 2013; 172(4): 447–458. Dostupné z DOI: <http://dx.doi.org/10.1007/s00431–012–1771-z>.
- Elias AF, Johnson MR, Boitnott JK et al. Neonatal cholestasis as initial manifestation of type 2 Gaucher disease: a continuum in the spectrum of early onset Gaucher disease. JIMD Rep 2012; 5: 95–98. Dostupné z DOI: <http://dx.doi.org/10.1007/8904_2011_104>.
- Park JK, Orvisky E, Tayebi N et al. Myoclonic epilepsy in Gaucher disease: genotype-phenotype insights from a rare patient subgroup. Pediatr Rep 2003; 53(3): 387–395. Dostupné z DOI: <http://dx.doi.org/10.1203/01.PDR.0000049515.79882.94>.
- Belmatoug N, Di Rocco M, Fraga C et al. Management and monitoring recommendations for the use of eliglustat in adults with type 1 Gaucher disease in Europe. Eur J Intern Med 2017; 37:25–32. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ejim.2016.07.011>.
- Waldek S, Feriozzi S. Fabry nephropathy: a review – how can we optimize the management of Fabry nephropathy? BMC Nephrol 2014; 15: 72. Dostupné z DOI: <http://dx.doi.org/10.1186/1471–2369–15–72>.
- Bacharova L, Ugander M. Left ventricular hypertrophy: The relationship between the electrocardiogram and cardiovascular magnetic resonance imaging. Ann Noninvasive Electrocardiol 2014; 19(6): 524–533. Dostupné z DOI: <http://dx.doi.org/10.1111/anec.12223>.
- Schiffmann R, Ries M. Fabry Disease: A Disorder of Childhood Onset. Pediatr Neurol 2016; 64: 10–20. Dostupné z DOI: <http://dx.doi.org/10.1016/j.pediatrneurol.2016.07.001>.
- Vanier MT. Niemann-Pick diseases. Handb Clin Neurol 2013; 113: 1717–1721. Dostupné z DOI: <http://dx.doi.org/10.1016/B978–0-444–59565–2.00041–1>.
- Mengel E, Klünemann HH, Lourenço CM et al. Niemann-Pick disease type C symptomatology: an expert-based clinical description. Orphanet J Rare Dis 2013; 8: 166. Dostupné z DOI: <http://dx.doi.org/10.1186/1750–1172–8-166>.
- Jahnova H, Dvorakova L, Vlaskova H et al. Observational, retrospective study of a large cohort of patients with Niemann-Pick disease type C in the Czech Republic: a surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J Rare Dis 2014; 9: 140. Dostupné z DOI: <http://dx.doi.org/10.1186/s13023–014–0140–6>.
- Hoeg JM, Demosky SJ, Pescovitz OH et al. Cholesteryl ester storage disease and Wolman disease: phenotypic variants of lysosomal acid cholesteryl ester hydrolase deficiency. Am J Hum Genet 1984; 36(6): 1190–1203.
- Maciejko JJ. Managing Cardiovascular Risk in Lysosomal Acid Lipase Deficiency. Am J Cardiovasc Drugs 2017; 17(3): 217–231. Dostupné z DOI: <http://dx.doi.org/10.1007/s40256–017–0216–5>. Erratum in Erratum to: Managing Cardiovascular Risk in Lysosomal Acid Lipase Deficiency. [Am J Cardiovasc Drugs. 2017].
- Erwin AL. The role of sebelipase alfa in the treatment of lysosomal acid lipase deficiency. Therap Adv Gastroenterol 2017; 10(7): 553–562. Dostupné z DOI: <http://dx.doi.org/10.1177/1756283X17705775>.
Štítky
Angiologie Diabetologie Interní lékařství Kardiologie Praktické lékařství pro dospěléČlánek vyšel v časopise
Athero Review

2018 Číslo 3
Nejčtenější v tomto čísle
- Fibráty v roce 2018: příběh pokračuje
- Mentálna anorexia a poruchy lipdového metabolizmu
- Statiny, mozek a velmi nízký cholesterol: jde to dohromady?
- Lyzozómové ochorenia ako príčina dyslipoproteinémie